Run scMODAL on the CITE-seq PBMC dataset
In this tutorial, we show how to run scMODAL integration of the CITE-seq PMBC data. The raw dataset is available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164378. Preprocessed data can be found here: https://drive.google.com/drive/folders/16MyDgz3iLKonZmshiU8YJxnBJw6UrGOx?usp=sharing.
Import packages
[1]:
import numpy as np
import pandas as pd
import scanpy as sc
import anndata as ad
import os
import scmodal
import warnings
warnings.filterwarnings("ignore")
Preprocessing
Load data
[2]:
adata_RNA = sc.read_h5ad('./data/citeseq_pbmc/multi.h5ad')
adata_RNA.var.index = adata_RNA.var['_index']
adata_RNA.X = adata_RNA.raw.X.toarray()
counts_ADT = pd.read_csv('./data/citeseq_pbmc/ADT.csv').T
adata_ADT = ad.AnnData(X = counts_ADT.values)
adata_ADT.obs.index = counts_ADT.index
adata_ADT.var.index = counts_ADT.columns
adata_ADT.obs = adata_RNA.obs.loc[adata_ADT.obs.index]
adata_RNA = adata_RNA[adata_RNA.obs.donor == 'P1']
adata_ADT = adata_ADT[adata_RNA.obs.index]
adata_RNA = adata_RNA[adata_RNA.obs['celltype.l2'].values != 'Doublet']
adata_ADT = adata_ADT[adata_ADT.obs['celltype.l2'].values != 'Doublet']
Normalization
[6]:
RNA_counts = RNA_shared.X.sum(axis=1)
ADT_counts = ADT_shared.X.sum(axis=1)
target_sum = np.maximum(np.median(RNA_counts.copy()), 20)
sc.pp.normalize_total(RNA_shared, target_sum=target_sum)
sc.pp.log1p(RNA_shared)
sc.pp.normalize_total(ADT_shared, target_sum=target_sum)
sc.pp.log1p(ADT_shared)
sc.pp.normalize_total(RNA_unshared)
sc.pp.log1p(RNA_unshared)
sc.pp.normalize_total(ADT_unshared)
sc.pp.log1p(ADT_unshared)
adata1 = ad.concat([RNA_shared, RNA_unshared], axis=1)
adata2 = ad.concat([ADT_shared, ADT_unshared], axis=1)
sc.pp.scale(adata1, max_value=10)
sc.pp.scale(adata2, max_value=10)
Running scMODAL
[7]:
model = scmodal.model.Model(model_path="./CITE-seq_PBMC")
model.preprocess(adata1, adata2, shared_gene_num=RNA_shared.shape[1])
[8]:
model.train()
model.eval()
Begining time: Wed Aug 28 09:37:54 2024
step 0, loss_D=0.155968, loss_GAN=-0.077693, loss_AE=419.729950, loss_Geo=-7.370721, loss_LA=5951.116211, loss_MNN=73.648102
step 2000, loss_D=1.370855, loss_GAN=-1.360073, loss_AE=9.889675, loss_Geo=-19.473022, loss_LA=0.170495, loss_MNN=0.169188
step 4000, loss_D=1.338295, loss_GAN=-1.323021, loss_AE=9.734790, loss_Geo=-19.494995, loss_LA=0.212534, loss_MNN=0.183071
step 6000, loss_D=1.314173, loss_GAN=-1.303403, loss_AE=9.588865, loss_Geo=-19.493710, loss_LA=0.104717, loss_MNN=0.169227
step 8000, loss_D=1.361248, loss_GAN=-1.354216, loss_AE=9.343693, loss_Geo=-19.495127, loss_LA=0.056516, loss_MNN=0.158181
Ending time: Wed Aug 28 09:58:05 2024
Training takes 1210.30 seconds
Begining time: Wed Aug 28 09:58:05 2024
Ending time: Wed Aug 28 09:58:05 2024
Evaluating takes 0.14 seconds
[9]:
adata_integrated = ad.AnnData(X=model.latent)
adata_integrated.obs = pd.concat([adata_RNA.obs, adata_ADT.obs])
adata_integrated.obs['modality'] = ['RNA'] * adata_RNA.shape[0] + ['ADT'] * adata_ADT.shape[0]
scmodal.utils.compute_umap(adata_integrated)
UMAP(angular_rp_forest=True, local_connectivity=1, metric='correlation', min_dist=0.3, n_neighbors=30, random_state=1234, repulsion_strength=1, verbose=True)
Wed Aug 28 09:58:05 2024 Construct fuzzy simplicial set
Wed Aug 28 09:58:05 2024 Finding Nearest Neighbors
Wed Aug 28 09:58:05 2024 Building RP forest with 14 trees
Wed Aug 28 09:58:08 2024 NN descent for 15 iterations
1 / 15
2 / 15
3 / 15
Stopping threshold met -- exiting after 3 iterations
Wed Aug 28 09:58:26 2024 Finished Nearest Neighbor Search
Wed Aug 28 09:58:28 2024 Construct embedding
completed 0 / 200 epochs
completed 20 / 200 epochs
completed 40 / 200 epochs
completed 60 / 200 epochs
completed 80 / 200 epochs
completed 100 / 200 epochs
completed 120 / 200 epochs
completed 140 / 200 epochs
completed 160 / 200 epochs
completed 180 / 200 epochs
Wed Aug 28 09:59:04 2024 Finished embedding
[10]:
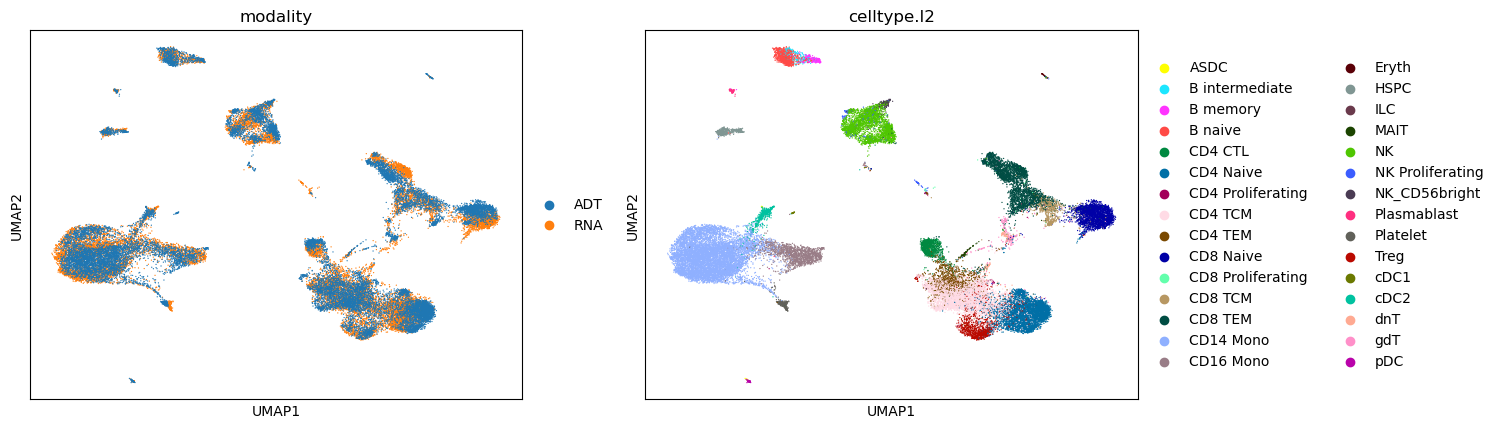
sc.pl.umap(adata_integrated, color=['modality', 'celltype.l2'])
Evaluation
[11]:
from scipy.spatial.distance import cdist
dist_mtx = cdist(model.latent[adata1.shape[0]:, :],
model.latent[:adata1.shape[0], :],
metric='euclidean') # Transfer labels from RNA to ADT
matching = dist_mtx.argsort()[:, :1]
df1_labels = adata_RNA.obs["celltype.l1"].values
df2_labels = adata_ADT.obs["celltype.l1"].values
print("Label transfer accuracy: ", np.sum(df1_labels == df2_labels[matching.reshape(-1)]) / adata_RNA.shape[0])
Label transfer accuracy: 0.9782078296550959
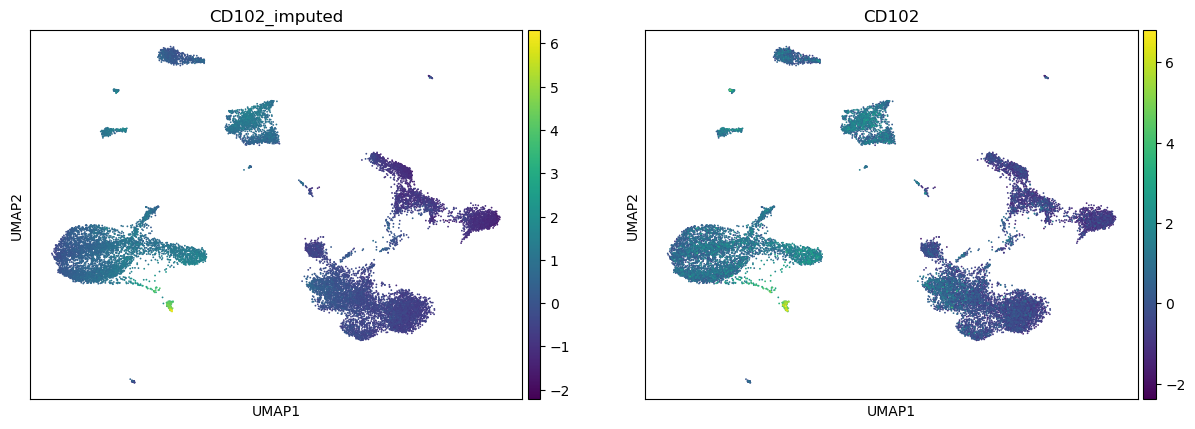
Feature imputation
In this example, we show the utility of scMODAL to impute protein abundance levels based on gene expressions.
[12]:
# get imputed features in DataFrame
model.get_imputed_df()
imputed_df = model.imputed_df_AtoB
# get ground truth for comparison
true_df = pd.DataFrame(adata2.X[:, :RNA_shared.shape[1]], index=adata2.obs.index, columns=adata2.var.feature_name[:RNA_shared.shape[1]])
true_df = true_df.groupby(true_df.columns, axis=1).mean()
[17]:
adata_imputed = ad.AnnData(X=np.concatenate([imputed_df.values, true_df.values], axis=1))
adata_imputed.obs = adata_RNA.obs
adata_imputed.var.index = list(imputed_df.columns + '_imputed') + list(true_df.columns)
adata_imputed.obsm['X_umap'] = adata_integrated.obsm['X_umap'][:adata_RNA.shape[0]]
[18]:
# plot imputed protein abundance levels vs ground truth
sc.pl.umap(adata_imputed, color=['CD102_imputed', 'CD102'])