Run scMODAL on the TEA-seq PBMC dataset
In this tutorial, we apply scMODAL to the tri-modality integration of the TEA-seq data. The raw dataset is available at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158013. Preprocessed data can be found here: https://drive.google.com/drive/folders/1HT7dTxd3AME-9fbXiO2cLQ_1zZnXA7AF?usp=sharing, where ATAC data were transfered to the gene activity matrix using Signac.
Import packages
[1]:
import numpy as np
import pandas as pd
import scanpy as sc
import anndata as ad
import os
import scmodal
import warnings
warnings.filterwarnings("ignore")
Preprocessing
Load data
[2]:
adata_RNA = sc.read_h5ad('./data/tea-seq/RNA.h5ad')
adata_ATAC = sc.read_h5ad('./data/tea-seq/ATAC.h5ad')
adata_ADT = sc.read_h5ad('./data/tea-seq/ADT.h5ad')
Normalization
[5]:
RNA_counts = RNA_shared.X.sum(axis=1)
ADT_counts = ADT_shared.X.sum(axis=1)
target_sum = np.maximum(np.median(np.array(RNA_counts).copy()), 20)
sc.pp.normalize_total(RNA_shared, target_sum=target_sum)
sc.pp.log1p(RNA_shared)
sc.pp.normalize_total(ADT_shared, target_sum=target_sum)
sc.pp.log1p(ADT_shared)
sc.pp.normalize_total(RNA_unshared)
sc.pp.log1p(RNA_unshared)
sc.pp.normalize_total(ADT_unshared)
sc.pp.log1p(ADT_unshared)
adata1 = ad.concat([ADT_shared, ADT_unshared], axis=1)
adata2 = ad.concat([RNA_shared, RNA_unshared], axis=1)
sc.pp.scale(adata1, max_value=10)
sc.pp.scale(adata2, max_value=10)
sc.pp.highly_variable_genes(adata_ATAC, flavor='seurat_v3', n_top_genes=1000)
adata_ATAC = adata_ATAC[:, adata_ATAC.var.highly_variable].copy()
adata_ATAC.var['feature_name'] = adata_ATAC.var.index.values
adata3 = adata_ATAC.copy()
sc.pp.normalize_total(adata3)
sc.pp.log1p(adata3)
sc.pp.scale(adata3, max_value=10)
[6]:
RNA_ATAC_shared = sorted(list(adata2.var.index & adata3.var.index))
adata23_shared = ad.concat([adata2[:, RNA_ATAC_shared], adata3[:, RNA_ATAC_shared]])
sc.tl.pca(adata23_shared, n_comps=30)
Running scMODAL
[7]:
model = scmodal.model.Model(training_steps=20000, model_path="./TEA-seq_PBMC")
[8]:
model.integrate_datasets_feats(input_feats=[adata1.X, adata2.X, adata3.X],
paired_input_MNN=[[adata1.X[:, :RNA_shared.shape[1]], adata2.X[:, :RNA_shared.shape[1]]],
[adata23_shared.obsm['X_pca'][:adata2.shape[0]], adata23_shared.obsm['X_pca'][adata2.shape[0]:]]])
Begining time: Tue Oct 1 14:44:44 2024
step 0, loss_D=0.343220, loss_GAN=-0.194079, loss_AE=278.313232, loss_Geo=-11.916582, loss_LA=9018.402344, loss_MNN=70.105209
step 2000, loss_D=2.316036, loss_GAN=-2.241776, loss_AE=16.127258, loss_Geo=-29.139099, loss_LA=0.728096, loss_MNN=0.446115
step 4000, loss_D=2.310611, loss_GAN=-2.253766, loss_AE=15.033018, loss_Geo=-29.182095, loss_LA=0.527380, loss_MNN=0.368130
step 6000, loss_D=2.656654, loss_GAN=-2.594337, loss_AE=15.177022, loss_Geo=-29.202566, loss_LA=0.473842, loss_MNN=0.321554
step 8000, loss_D=2.529740, loss_GAN=-2.481193, loss_AE=14.962239, loss_Geo=-29.191803, loss_LA=0.320206, loss_MNN=0.347442
step 10000, loss_D=2.273712, loss_GAN=-2.229443, loss_AE=14.667961, loss_Geo=-29.226091, loss_LA=0.312849, loss_MNN=0.357043
step 12000, loss_D=2.512218, loss_GAN=-2.470063, loss_AE=14.772261, loss_Geo=-29.217646, loss_LA=0.261980, loss_MNN=0.348535
step 14000, loss_D=2.609316, loss_GAN=-2.528668, loss_AE=15.029343, loss_Geo=-29.220142, loss_LA=0.373776, loss_MNN=0.394483
step 16000, loss_D=2.506408, loss_GAN=-2.464864, loss_AE=14.867597, loss_Geo=-29.198139, loss_LA=0.269569, loss_MNN=0.350754
step 18000, loss_D=2.559177, loss_GAN=-2.501715, loss_AE=14.843930, loss_Geo=-29.228626, loss_LA=0.237626, loss_MNN=0.382120
Ending time: Tue Oct 1 15:36:28 2024
Training takes 3103.66 seconds
Begining time: Tue Oct 1 15:36:28 2024
Ending time: Tue Oct 1 15:36:28 2024
Evaluating takes -0.00 seconds
[14]:
adata_integrated = ad.AnnData(X=model.latent)
adata_integrated.obs = pd.concat([adata_ADT.obs, adata_RNA.obs, adata_ATAC.obs])
adata_integrated.obs['modality'] = ['ADT'] * adata_ADT.shape[0] + ['RNA'] * adata_RNA.shape[0] + ['ATAC'] * adata_RNA.shape[0]
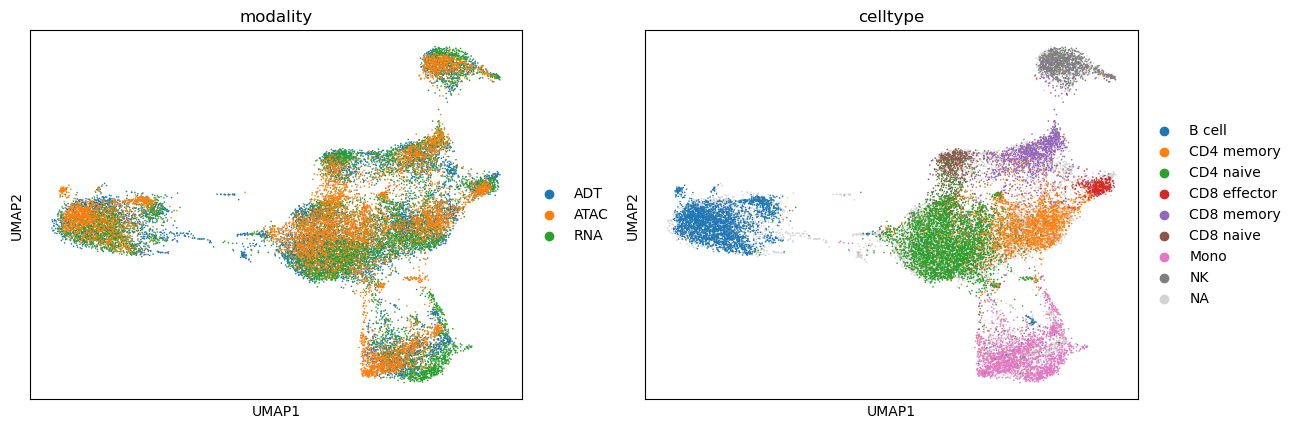
scmodal.utils.compute_umap(adata_integrated)
UMAP(angular_rp_forest=True, local_connectivity=1, metric='correlation', min_dist=0.3, n_neighbors=30, random_state=1234, repulsion_strength=1, verbose=True)
Tue Oct 1 15:38:18 2024 Construct fuzzy simplicial set
Tue Oct 1 15:38:18 2024 Finding Nearest Neighbors
Tue Oct 1 15:38:18 2024 Building RP forest with 12 trees
Tue Oct 1 15:38:18 2024 NN descent for 14 iterations
1 / 14
2 / 14
3 / 14
4 / 14
Stopping threshold met -- exiting after 4 iterations
Tue Oct 1 15:38:20 2024 Finished Nearest Neighbor Search
Tue Oct 1 15:38:20 2024 Construct embedding
completed 0 / 200 epochs
completed 20 / 200 epochs
completed 40 / 200 epochs
completed 60 / 200 epochs
completed 80 / 200 epochs
completed 100 / 200 epochs
completed 120 / 200 epochs
completed 140 / 200 epochs
completed 160 / 200 epochs
completed 180 / 200 epochs
Tue Oct 1 15:38:42 2024 Finished embedding
[16]:
sc.pl.umap(adata_integrated, color=['modality', 'celltype'])