Run scMODAL for the integration of tonsil datasets
In this tutorial, we apply scMODAL to the integration of tonsil CODEX, scRNA-seq and scATAC-seq datasets. The raw datasets are available at https://datadryad.org/stash/share/1OQtxew0Unh3iAdP-ELew-ctwuPTBz6Oy8uuyxqliZk, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE165860 and https://zenodo.org/records/11355186. Preprocessed data can be found here: https://drive.google.com/drive/folders/1-8Di5xv5hXqG68rOj4jAhBWEznt5oVnR?usp=sharing, where ATAC data were transfered to the gene activity matrix using Signac.
Import packages
[1]:
import numpy as np
import pandas as pd
import scanpy as sc
import anndata as ad
import commot as ct
import os
import scmodal
import warnings
warnings.filterwarnings("ignore")
Preprocessing
Load data
[2]:
adata_CODEX = ad.read_h5ad('./data/tonsil/adata_codex.h5ad')
adata_RNA = ad.read_h5ad('./data/tonsil/adata_rna.h5ad')
adata_ATAC = ad.read_h5ad('./data/tonsil/adata_atac.h5ad')
Normalization
[11]:
sc.pp.normalize_total(adata_RNA_shared, target_sum=np.median((np.exp(adata_CODEX_shared.X)-1).sum(axis=1)))
sc.pp.log1p(adata_RNA_shared)
sc.pp.normalize_total(adata_RNA_unshared)
sc.pp.log1p(adata_RNA_unshared)
adata_RNA = ad.concat([adata_RNA_shared, adata_RNA_unshared], axis=1)
adata_RNA.obs["celltype"] = adata_RNA_shared.obs["celltype"]
adata_CODEX = adata_CODEX_shared # CODEX data do not contain unlinked features with RNA data
adata_CODEX.obs["celltype"] = adata_CODEX.obs["celltype"]
sc.pp.scale(adata_RNA, max_value=10)
sc.pp.scale(adata_CODEX, max_value=10)
sc.pp.normalize_total(adata_ATAC_shared, target_sum=np.median((np.exp(adata_CODEX_ATAC_shared.X)-1).sum(axis=1)))
sc.pp.log1p(adata_ATAC_shared)
sc.pp.normalize_total(adata_ATAC_unshared)
sc.pp.log1p(adata_ATAC_unshared)
adata_ATAC = ad.concat([adata_ATAC_shared, adata_ATAC_unshared], axis=1)
adata_ATAC.obs["dataset"] = "ATAC"
adata_ATAC.obs["celltype"] = adata_ATAC_shared.obs["celltype"]
sc.pp.scale(adata_ATAC, max_value=10)
RNA_ATAC_shared = sorted(list(adata_RNA.var.index & adata_ATAC.var.index))
adata_CODEX.obs['modality'] = 'CODEX'
adata_RNA.obs['modality'] = 'RNA'
adata_ATAC.obs['modality'] = 'ATAC'
adata_RNA_ATAC_shared = ad.concat([adata_RNA[:, RNA_ATAC_shared], adata_ATAC[:, RNA_ATAC_shared]])
sc.tl.pca(adata_RNA_ATAC_shared, n_comps=30)
Running scMODAL
[12]:
model = scmodal.model.Model(training_steps=10000, lambdaMNN=5, lambdaGAN=0.5, model_path="./tonsil_tutorial")
model.integrate_datasets_feats(input_feats=[adata_CODEX.X, adata_RNA.X, adata_ATAC.X],
paired_input_MNN=[[adata_CODEX.X[:, :adata_RNA_shared.shape[1]], adata_RNA.X[:, :adata_RNA_shared.shape[1]]],
[adata_RNA_ATAC_shared.obsm['X_pca'][:adata_RNA.shape[0]], adata_RNA_ATAC_shared.obsm['X_pca'][adata_RNA.shape[0]:]], ])
Begining time: Mon Apr 28 21:07:28 2025
step 0, loss_D=0.896587, loss_GAN=-0.473515, loss_AE=276.851959, loss_Geo=-11.990168, loss_LA=10724.443359, loss_MNN=371.120636
step 2000, loss_D=2.224448, loss_GAN=-2.179823, loss_AE=15.710739, loss_Geo=-29.124660, loss_LA=0.176542, loss_MNN=0.530770
step 6000, loss_D=2.559398, loss_GAN=-2.521484, loss_AE=14.851513, loss_Geo=-29.160221, loss_LA=0.121875, loss_MNN=0.404307
step 8000, loss_D=2.610829, loss_GAN=-2.585304, loss_AE=14.858552, loss_Geo=-29.035725, loss_LA=0.094044, loss_MNN=0.401230
Ending time: Mon Apr 28 21:30:23 2025
Training takes 1374.56 seconds
Begining time: Mon Apr 28 21:30:23 2025
Ending time: Mon Apr 28 21:30:23 2025
Evaluating takes -0.00 seconds
[13]:
adata_integrated = ad.AnnData(X=model.latent)
adata_integrated.obs['modality'] = ['CODEX'] * adata_CODEX.shape[0] + ['RNA'] * adata_RNA.shape[0] + ['ATAC'] * adata_ATAC.shape[0]
adata_integrated.obs['celltype'] = list(adata_CODEX.obs['celltype'].values) + list(adata_RNA.obs['celltype'].values) + list(adata_ATAC.obs['celltype'].values)
scmodal.utils.compute_umap(adata_integrated)
UMAP(angular_rp_forest=True, local_connectivity=1, metric='correlation', min_dist=0.3, n_neighbors=30, random_state=1234, repulsion_strength=1, verbose=True)
Mon Apr 28 21:30:23 2025 Construct fuzzy simplicial set
Mon Apr 28 21:30:23 2025 Finding Nearest Neighbors
Mon Apr 28 21:30:23 2025 Building RP forest with 21 trees
Mon Apr 28 21:30:26 2025 NN descent for 17 iterations
1 / 17
2 / 17
3 / 17
4 / 17
Stopping threshold met -- exiting after 4 iterations
Mon Apr 28 21:32:23 2025 Finished Nearest Neighbor Search
Mon Apr 28 21:32:26 2025 Construct embedding
completed 0 / 200 epochs
completed 20 / 200 epochs
completed 40 / 200 epochs
completed 60 / 200 epochs
completed 80 / 200 epochs
completed 100 / 200 epochs
completed 120 / 200 epochs
completed 140 / 200 epochs
completed 160 / 200 epochs
completed 180 / 200 epochs
Mon Apr 28 21:34:25 2025 Finished embedding
[14]:
sc.pl.umap(adata_integrated, color=['modality', "celltype"])
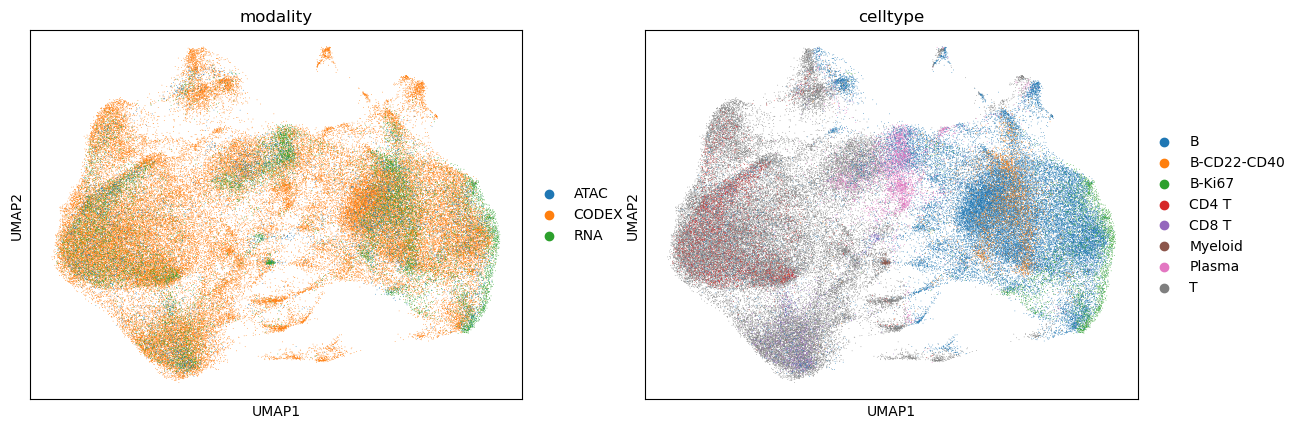
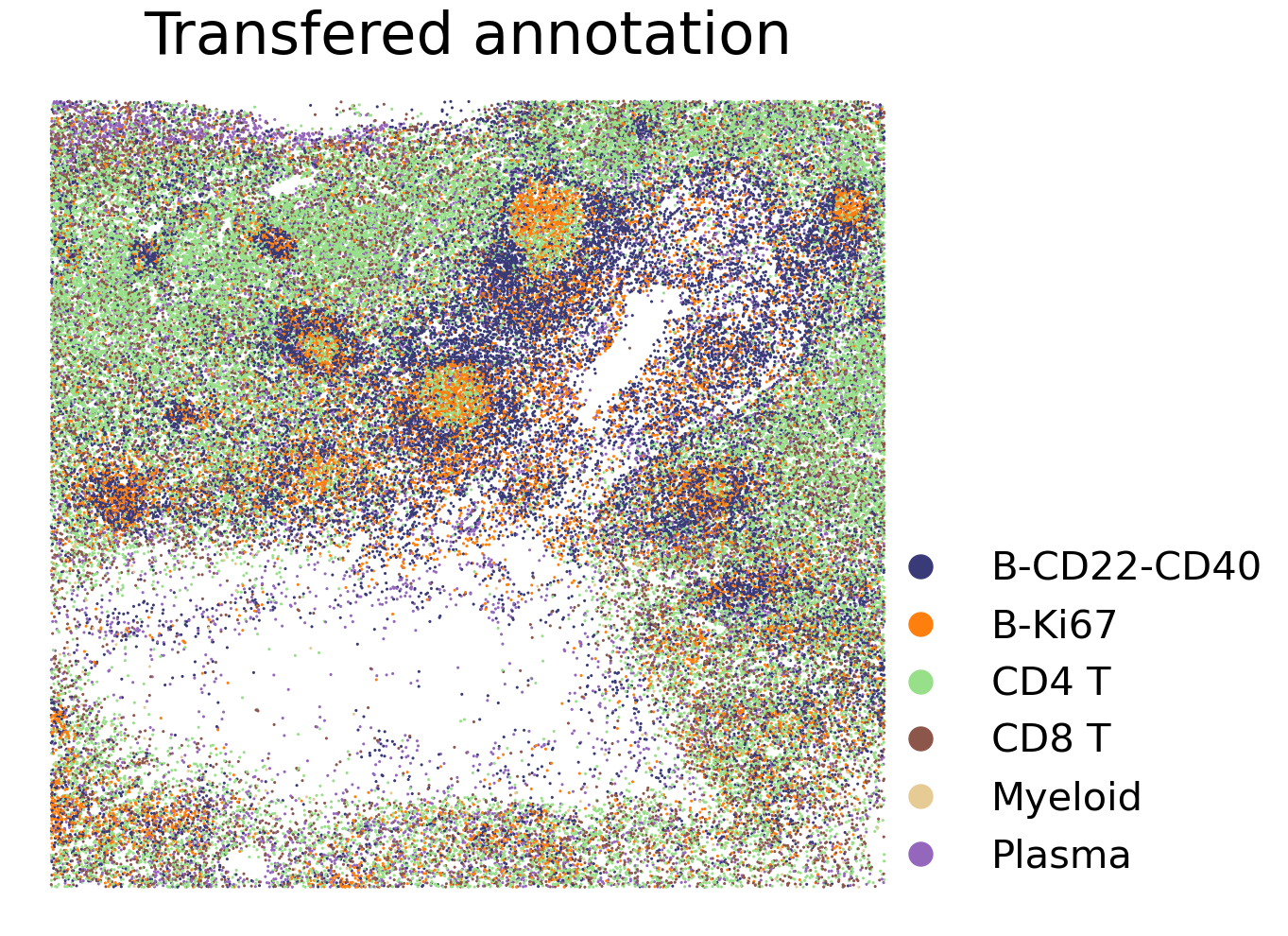
Label transfer from scRNA-seq to CODEX
[15]:
from scipy.spatial.distance import cdist
dist_mtx = cdist(model.latent[:adata_CODEX.shape[0], :],
model.latent[adata_CODEX.shape[0]:(adata_RNA.shape[0]+adata_CODEX.shape[0]), :],
metric='euclidean')
matching_all = dist_mtx.argsort()
matching = dist_mtx.argsort()[:, :1]
[16]:
from scipy import stats
n = 1
result = stats.mode(adata_RNA.obs["celltype"].values.astype(str)[matching_all[:, :n]], axis=1)
transfered = result[0].reshape(-1)
[17]:
from sklearn import preprocessing
colours = ListedColormap(['#393b79', '#ff7f0e', '#98df8a', '#8c564b', '#e7cb94', "tab:purple"])
le = preprocessing.LabelEncoder()
le.fit(sorted(set(transfered)))
label = le.fit_transform(transfered)
f = plt.figure(figsize=(10,10))
ax1 = f.add_subplot(1,1,1)
scatter1 = ax1.scatter(adata_CODEX.obsm['spatial'][:, 0], adata_CODEX.obsm['spatial'][:, 1], c=label, cmap=colours, label=transfered, s = 1.5, rasterized=True)
ax1.tick_params(axis='both',bottom=False, top=False, left=False, right=False, labelleft=False, labelbottom=False, grid_alpha=0)
l1 = f.legend(handles=scatter1.legend_elements()[0], labels=sorted(set(transfered)), loc="upper left", bbox_to_anchor=(0.9, 0.45),
markerscale=3., title_fontsize=30, fontsize=30, frameon=False, ncol=1)
l1._legend_box.align = "left"
ax1.set_title("Transfered annotation", fontsize=45)
f.tight_layout()
ax1.axis('off')
plt.show()
[ ]: